
在世界各國醫學論文中“運動神經元病” (MND)用以敘述關鍵為運動神經元或前角體細胞危害的一組或多個不一樣的病症。假如各種各樣名字的使用人不確立界定,常易搞混。有時候運動神經元病一詞歸納了全部的前角體細胞病症,包含寶寶型或青年人型脊神經性肌肉萎縮症和關鍵為上運動神經元危害的皮層脊神經轉性。而有時候它僅代表成年人病發的、原發性的、造成了巨大的前角體細胞病症。

肌肉萎縮側索硬底化綜合征的歸類:
運動神經元病的特性是漸變性肌肉僵硬和委縮(肌肉日漸消瘦)。可出現在一切年紀的成人。有幾種種類的運動神經元病,現階段廣泛認可的見解覺得肌肉萎縮側索硬底化(ALS),特發性肌肉萎縮(PMA)和特發性延髓性麻木(PBP)是一種病症的不一樣亞型,而另一型原發性側索硬底化(PLS)較為少見,須由驗屍確認,也可歸入運動神經元病。
假如僅有上運動神經元損傷,就稱之為原發性側索硬化症。
假如僅有下運動神經元損傷,就稱之為脊神經性肌肉萎縮或特發性肌肉萎縮,脊神經性肌肉萎縮是一種常染色體潛在性遺傳疾病,少年兒童病發多,成年人後病發少,近端對稱乏力顯著。特發性肌肉萎縮多見成年人發病,病發不一樣,遠端及遠側均可病發。一些創作者覺得特發性肌肉萎縮不管從臨床症狀及預後都能夠當做一個單獨的病症,但其與肌肉萎縮側索硬底化電生理的主要表現差別之小,常無法區別。並且假如對特發性肌肉萎縮症或特發性延髓性麻木進一步查驗便會發覺他們也存有上運動神經元的損害,驗屍證實僅有下運動神經元危害的特發性肌肉萎縮非常少。近年來的科學研究又梳理出很多獨特種類如連枷臂綜合症、連枷腿綜合症等,但趨向因此肌肉萎縮側索硬底化亞型,是具備不一樣臨床醫學燃氣表的同一病症。
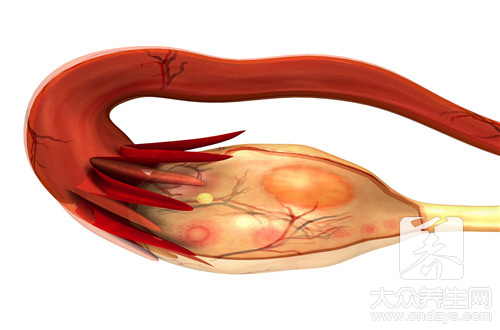
假如上、下運動神經元均損傷,就稱之為肌肉萎縮側索硬底化,它是運動神經元病最普遍的方式。
更有一些醫師用“運動神經元病症”(MNDs)指除原來MND之外的別的關鍵危害運動神經的病症如痙攣性截癱,平山病、肯尼迪病及其脊神經性肌肉萎縮(SMA)。
現階段中國歸類未有統一的了解,一般參考國外文獻的一些分類方法。我們趨向用運動神經元病症這一界定範圍廣的病症專有名詞下開展實際歸類。
文中提及的運動神經元病和肌肉萎縮側索硬底化是同一病症,後麵一種按國際性神經病學同盟1998年診斷標準確診,有可能包含經典型性肌肉萎縮側索硬底化、特發性肌肉萎縮、特發性延髓(球)麻木及原發性側索硬底化。
運動神經元病症包含了危害運動神經元的病症全部別的病症,如包含肌肉萎縮側索硬底化、特發性肌肉萎縮、原發性側索硬底化、特發性延髓(球)麻木、脊神經性肌肉萎縮、痙攣性截癱、多灶性運動精神病、平山病及肯尼迪病等各種類型。
肌肉萎縮側索硬底化中最普遍的種類是散發性肌肉萎縮側索硬底化。此外大概有20%的病人與基因遺傳相關,歸屬於大家族性肌肉萎縮側索硬底化。在西太平洋地區(關島、日本國的Kii半島花園、巴布亞新幾內亞)曾發現異常的高患病率。對關島肌肉萎縮側索硬底化病人的進一步科學研究結果顯示高患病率是因為飲食搭配中帶有獨特的有毒物質(如鋁、蘇鐵素)。伴隨著這一地域的西方化,所依靠的傳統式食品降低,肌肉萎縮側索硬底化的患病率出現減少的發展趨勢。










